Creating A Transcriptome Map
Overview
Teaching: 30 min
Exercises: 30 minQuestions
How do I create and interpret a transcriptome map?
Objectives
Describe a transcriptome map.
Interpret a transcriptome map.
Load Libraries
library(tidyverse)
library(qtl2)
library(qtl2convert)
library(GGally)
library(broom)
library(corrplot)
library(RColorBrewer)
library(qtl2ggplot)
source("../code/gg_transcriptome_map.R")
source("../code/qtl_heatmap.R")
Load Data
#expression data
load("../data/attie_DO500_expr.datasets.RData")
##mapping data
load("../data/attie_DO500_mapping.data.RData")
##genoprobs
probs = readRDS("../data/attie_DO500_genoprobs_v5.rds")
##phenotypes
load("../data/attie_DO500_clinical.phenotypes.RData")
expr.mrna <- norm
In this lesson we are going to learn how to create a transcriptome map. At transcriptome map shows the location of the eQTL peak compared its gene location, giving information about cos and trans eQTLs.
We are going to use the file that we created in the previous lesson with the lod scores greater than 6: gene.norm_qtl_peaks_cis.trans.csv.
You can load it in using the following code:
lod_summary = read.csv("../results/gene.norm_qtl_peaks_cis.trans.csv")
lod_summary$marker.id <- paste0(lod_summary$chr,
"_",
lod_summary$pos * 1000000)
ensembl = get_ensembl_genes()
snapshotDate(): 2022-04-25
loading from cache
Importing File into R ..
id = ensembl$gene_id
chr = seqnames(ensembl)
start = start(ensembl) * 1e-6
end = end(ensembl) * 1e-6
df = data.frame(ensembl = id,
gene_chr = chr,
gene_start = start,
gene_end = end,
stringsAsFactors = F)
colnames(lod_summary)[colnames(lod_summary) == "lodcolumn"] = "ensembl"
colnames(lod_summary)[colnames(lod_summary) == "chr"] = "qtl_chr"
colnames(lod_summary)[colnames(lod_summary) == "pos"] = "qtl_pos"
colnames(lod_summary)[colnames(lod_summary) == "lod"] = "qtl_lod"
lod_summary = left_join(lod_summary, df, by = "ensembl")
lod_summary = mutate(lod_summary,
gene_chr = factor(gene_chr, levels = c(1:19, "X")),
qtl_chr = factor(qtl_chr, levels = c(1:19, "X")))
rm(df)
We can summarise which eQTLs are cis or trans. Remember a cis qtl is… and trans eqtl is….
lod_summary$cis.trans <- ifelse(lod_summary$qtl_chr == lod_summary$gene_chr, "cis", "trans")
table(lod_summary$cis.trans)
cis trans
47 65
Plot Transcriptome Map
lod_summary = mutate(lod_summary, cis = (gene_chr == qtl_chr) & (abs(gene_start - qtl_pos) < 4))
out.plot = ggtmap(data = lod_summary %>% filter(qtl_lod >= 7.18), cis.points = TRUE, cis.radius = 4)
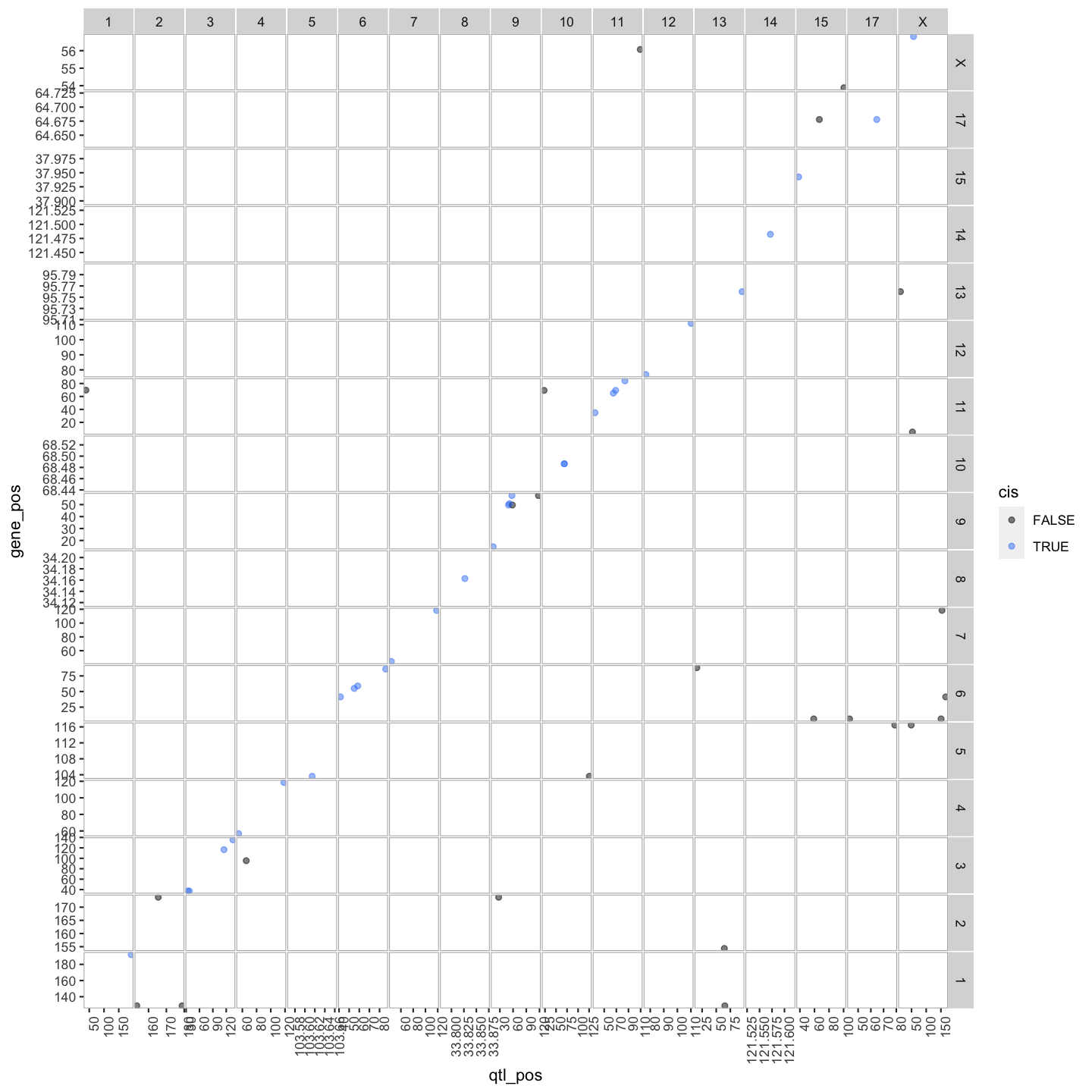
pdf("../results/transcriptome_map_cis.trans.pdf", width = 10, height = 10)
out.plot
dev.off()
quartz_off_screen
2
out.plot
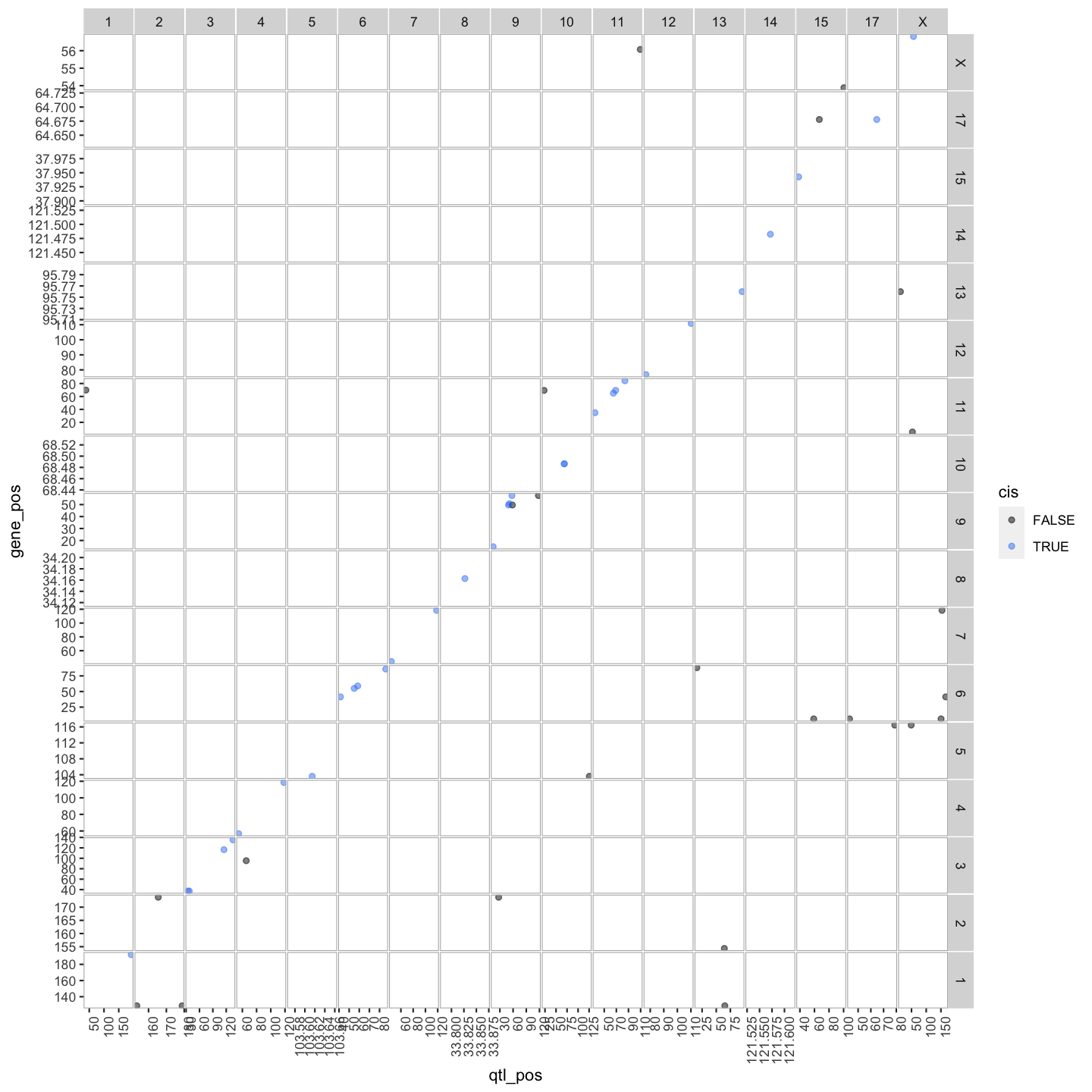
Key Points
Transcriptome maps aid in understanding gene expression regulation.