Transcriptome Map of cis and trans eQTL
Overview
Teaching: 10 min
Exercises: 20 minQuestions
How do I create a full transcriptome map?
Objectives
Load Libraries
library(tidyverse)
library(qtl2)
library(qtl2convert)
#library(qtl2db)
library(GGally)
library(broom)
library(knitr)
library(corrplot)
library(RColorBrewer)
library(qtl2ggplot)
source("../code/gg_transcriptome_map.R")
source("../code/qtl_heatmap.R")
Load Data
#expression data
load("../data/attie_DO500_expr.datasets.RData")
##loading previous results
load("../data/dataset.islet.rnaseq.RData")
##mapping data
load("../data/attie_DO500_mapping.data.RData")
##genoprobs
probs = readRDS("../data/attie_DO500_genoprobs_v5.rds")
##phenotypes
load("../data/attie_DO500_clinical.phenotypes.RData")
lod_summary = dataset.islet.rnaseq$lod.peaks
ensembl = get_ensembl_genes()
id = ensembl$gene_id
chr = seqnames(ensembl)
start = start(ensembl) * 1e-6
end = end(ensembl) * 1e-6
df = data.frame(ensembl = id, gene_chr = chr, gene_start = start, gene_end = end,
stringsAsFactors = F)
colnames(lod_summary)[colnames(lod_summary) == "annot.id"] = "ensembl"
colnames(lod_summary)[colnames(lod_summary) == "chrom"] = "qtl_chr"
colnames(lod_summary)[colnames(lod_summary) == "pos"] = "qtl_pos"
colnames(lod_summary)[colnames(lod_summary) == "lod"] = "qtl_lod"
lod_summary = left_join(lod_summary, df, by = "ensembl")
lod_summary = mutate(lod_summary, gene_chr = factor(gene_chr, levels = c(1:19, "X")),
qtl_chr = factor(qtl_chr, levels = c(1:19, "X")))
rm(df)
## summary:
lod_summary$cis.trans <- ifelse(lod_summary$qtl_chr == lod_summary$gene_chr, "cis", "trans")
table(lod_summary$cis.trans)
cis trans
14549 24299
Plot Transcriptome Map
lod_summary = mutate(lod_summary, cis = (gene_chr == qtl_chr) & (abs(gene_start - qtl_pos) < 4))
out.plot = ggtmap(data = lod_summary %>% filter(qtl_lod >= 7.18), cis.points = TRUE, cis.radius = 4)
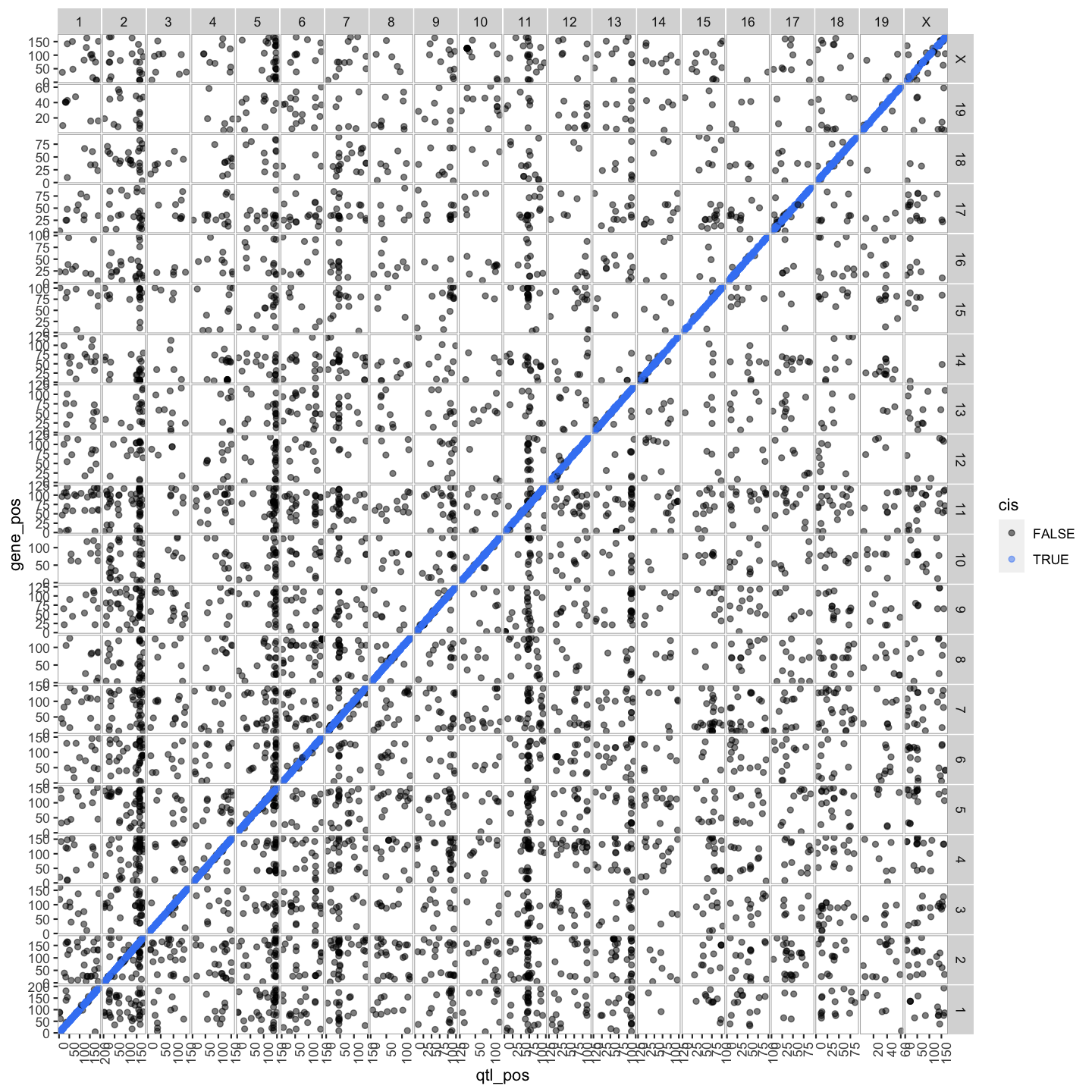
out.plot
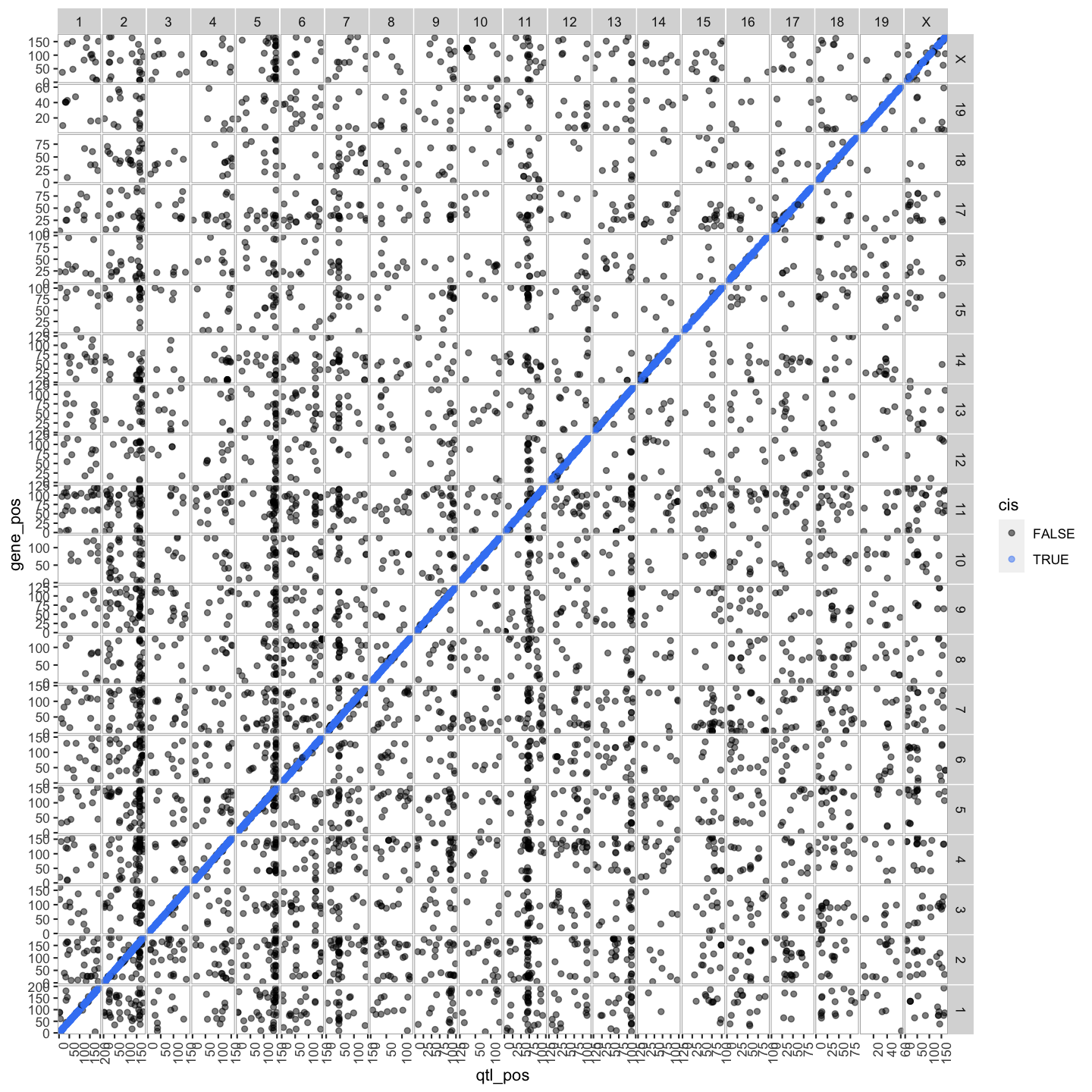
QTL Density Plot
breaks = matrix(c(seq(0, 200, 4), seq(1, 201, 4), seq(2, 202, 4), seq(3, 203, 4)), ncol = 4)
tmp = as.list(1:ncol(breaks))
for(i in 1:ncol(breaks)) {
tmp[[i]] = lod_summary %>%
filter(qtl_lod >= 7.18 & cis == FALSE) %>%
arrange(qtl_chr, qtl_pos) %>%
group_by(qtl_chr) %>%
mutate(win = cut(qtl_pos, breaks = breaks[,i])) %>%
group_by(qtl_chr, win) %>%
summarize(cnt = n()) %>%
separate(win, into = c("other", "prox", "dist")) %>%
mutate(prox = as.numeric(prox),
dist = as.numeric(dist),
mid = 0.5 * (prox + dist)) %>%
dplyr::select(qtl_chr, mid, cnt)
}
trans = bind_rows(tmp[[1]], tmp[[1]], tmp[[3]], tmp[[4]])
rm(tmp)
out.plot = ggplot(trans, aes(mid, cnt)) +
geom_line() +
geom_hline(aes(yintercept = 100), linetype = 2, color = "grey50") +
facet_grid(.~qtl_chr, scales = "free") +
theme(panel.background = element_blank(),
panel.border = element_rect(fill = 0, color = "grey70"),
panel.spacing = unit(0, "lines"),
axis.text.x = element_text(angle = 90)) +
labs(title = "trans-eQTL Histogram", x = "Mb", y = "Number of Transcripts")
out.plot
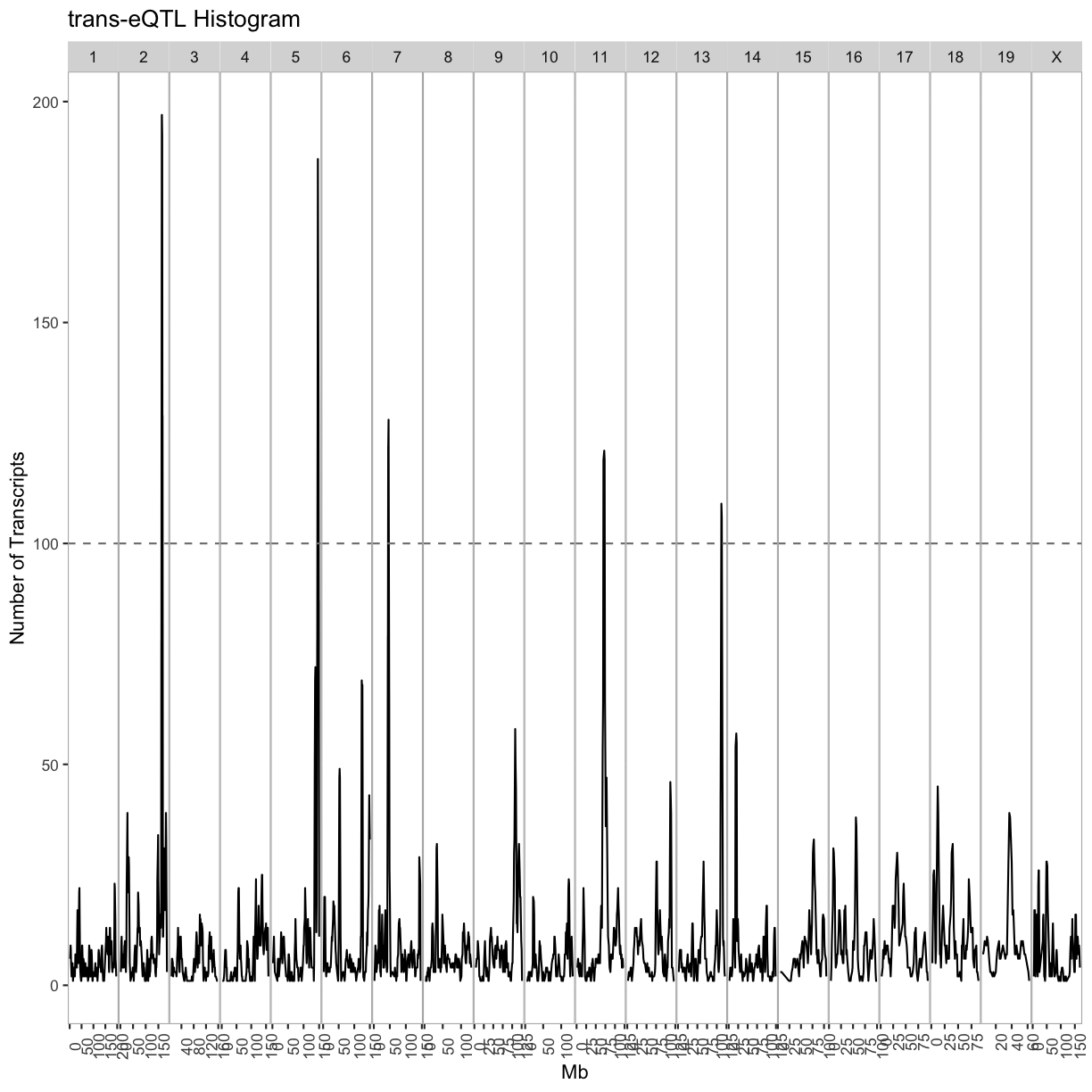
breaks = matrix(c(seq(0, 200, 4), seq(1, 201, 4), seq(2, 202, 4), seq(3, 203, 4)), ncol = 4)
tmp = as.list(1:ncol(breaks))
for(i in 1:ncol(breaks)) {
tmp[[i]] = lod_summary %>%
filter(qtl_lod >= 7.18 & cis == TRUE) %>%
arrange(qtl_chr, qtl_pos) %>%
group_by(qtl_chr) %>%
mutate(win = cut(qtl_pos, breaks = breaks[,i])) %>%
group_by(qtl_chr, win) %>%
summarize(cnt = n()) %>%
separate(win, into = c("other", "prox", "dist")) %>%
mutate(prox = as.numeric(prox),
dist = as.numeric(dist),
mid = 0.5 * (prox + dist)) %>%
dplyr::select(qtl_chr, mid, cnt)
}
cis = bind_rows(tmp[[1]], tmp[[2]], tmp[[3]], tmp[[4]])
rm(tmp1, tmp2, tmp3, tmp4)
out.plot = ggplot(cis, aes(mid, cnt)) +
geom_line(color = "#4286f4") +
geom_hline(aes(yintercept = 100), linetype = 2, color = "grey50") +
facet_grid(.~qtl_chr, scales = "free") +
theme(panel.background = element_blank(),
panel.border = element_rect(fill = 0, color = "grey70"),
panel.spacing = unit(0, "lines"),
axis.text.x = element_text(angle = 90)) +
labs(title = "cis-eQTL Histogram", x = "Mb", y = "Number of Transcripts")
out.plot
tmp = lod_summary %>%
filter(qtl_lod >= 7.18) %>%
group_by(cis) %>%
count()
kable(tmp, caption = "Number of cis- and trans-eQTL")
Table: Number of cis- and trans-eQTL
| cis | n |
|---|---|
| FALSE | 5617 |
| TRUE | 12677 |
| NA | 526 |
rm(tmp)
Islet RNASeq eQTL Hotspots
Select eQTL Hotspots
Select trans-eQTL hotspots with more than 100 genes at the 7.18 LOD thresholds. Retain the maximum per chromosome.
hotspots = trans %>%
group_by(qtl_chr) %>%
filter(cnt >= 100) %>%
summarize(center = median(mid)) %>%
mutate(proximal = center - 2, distal = center + 2)
kable(hotspots, caption = "Islet trans-eQTL hotspots")
Table: Islet trans-eQTL hotspots
| qtl_chr | center | proximal | distal |
|---|---|---|---|
| 2 | 165.5 | 163.5 | 167.5 |
| 5 | 146.0 | 144.0 | 148.0 |
| 7 | 46.0 | 44.0 | 48.0 |
| 11 | 71.0 | 69.0 | 73.0 |
| 13 | 112.5 | 110.5 | 114.5 |
cis.hotspots = cis %>%
group_by(qtl_chr) %>%
filter(cnt >= 100) %>%
summarize(center = median(mid)) %>%
mutate(proximal = center - 2, distal = center + 2)
kable(cis.hotspots, caption = "Islet cis-eQTL hotspots")
Table: Islet cis-eQTL hotspots
| qtl_chr | center | proximal | distal |
|---|---|---|---|
| 7 | 29.0 | 27.0 | 31.0 |
| 17 | 34.5 | 32.5 | 36.5 |
Given the hotspot locations, retain all genes with LOD > 7.18 and trans-eQTL within +/- 4Mb of the mid-point of the hotspot.
hotspot.genes = as.list(hotspots$qtl_chr)
names(hotspot.genes) = hotspots$qtl_chr
for(i in 1:nrow(hotspots)) {
hotspot.genes[[i]] = lod_summary %>%
filter(qtl_lod >= 7.18) %>%
filter(qtl_chr == hotspots$qtl_chr[i] &
qtl_pos >= hotspots$proximal[i] &
qtl_pos <= hotspots$distal[i] &
(gene_chr != hotspots$qtl_chr[i] |
(gene_chr == hotspots$qtl_chr[i] &
gene_start > hotspots$distal[i] + 1 &
gene_end < hotspots$proximal[i] - 1)))
write_csv(hotspot.genes[[i]], file = paste0("../results/chr", names(hotspot.genes)[i], "_hotspot_genes.csv"))
}
Number of genes in each hotspot.
hotspots = data.frame(hotspots, count = sapply(hotspot.genes, nrow))
kable(hotspots, caption = "Number of genes per hotspot")
Table: Number of genes per hotspot
| qtl_chr | center | proximal | distal | count | |
|---|---|---|---|---|---|
| 2 | 2 | 165.5 | 163.5 | 167.5 | 147 |
| 5 | 5 | 146.0 | 144.0 | 148.0 | 182 |
| 7 | 7 | 46.0 | 44.0 | 48.0 | 123 |
| 11 | 11 | 71.0 | 69.0 | 73.0 | 126 |
| 13 | 13 | 112.5 | 110.5 | 114.5 | 104 |
cis.hotspot.genes = as.list(cis.hotspots$qtl_chr)
names(cis.hotspot.genes) = cis.hotspots$qtl_chr
for(i in 1:nrow(cis.hotspots)) {
cis.hotspot.genes[[i]] = lod_summary %>%
dplyr::select(ensembl, marker.id, qtl_chr, qtl_pos, qtl_lod) %>%
filter(qtl_lod >= 7.18) %>%
filter(qtl_chr == cis.hotspots$qtl_chr[i] &
qtl_pos >= cis.hotspots$proximal[i] &
qtl_pos <= cis.hotspots$distal[i])
write_csv(cis.hotspot.genes[[i]], file = paste0("../results/chr", names(cis.hotspot.genes)[i], "_cis_hotspot_genes.csv"))
}
Number of genes in each cis-hotspot.
cis.hotspots = data.frame(cis.hotspots, count = sapply(cis.hotspot.genes, nrow))
kable(cis.hotspots, caption = "Number of genes per cis-hotspot")
Table: Number of genes per cis-hotspot
| qtl_chr | center | proximal | distal | count | |
|---|---|---|---|---|---|
| 7 | 7 | 29.0 | 27.0 | 31.0 | 120 |
| 17 | 17 | 34.5 | 32.5 | 36.5 | 188 |
Get the expression of genes that map to each hotspot.
for(i in 1:length(hotspot.genes)) {
tmp = data.frame(ensembl = hotspot.genes[[i]]$ensembl, t(expr.mrna[,hotspot.genes[[i]]$ensembl]))
hotspot.genes[[i]] = left_join(hotspot.genes[[i]], tmp, by = "ensembl")
write_csv(hotspot.genes[[i]], file = paste0("../results/chr", names(hotspot.genes)[i], "_hotspot_genes.csv"))
}
Error in h(simpleError(msg, call)): error in evaluating the argument 'x' in selecting a method for function 't': object 'expr.mrna' not found
Hotspot Gene Correlation
breaks = -100:100/100
colors = colorRampPalette(rev(brewer.pal(11, "Spectral")))(length(breaks) - 1)
for(i in 1:length(hotspot.genes)) {
chr = names(hotspot.genes)[i]
tmp = hotspot.genes[[i]] %>%
dplyr::select(starts_with("DO")) %>%
t() %>%
as.matrix() %>%
cor()
dimnames(tmp) = list(hotspot.genes[[i]]$ensembl, hotspot.genes[[i]]$ensembl)
side.colors = cut(hotspot.genes[[i]]$qtl_lod, breaks = 100)
side.colors = colorRampPalette(rev(brewer.pal(9, "YlOrRd")))(length(levels(side.colors)))[as.numeric(side.colors)]
names(side.colors) = rownames(tmp)
heatmap(tmp, symm = TRUE, scale = "none", main = paste("Chr", chr, "Gene Correlation"), breaks = breaks, col = colors, RowSideColors = side.colors, ColSideColors = side.colors)
}
Error in hclustfun(distfun(x)): NA/NaN/Inf in foreign function call (arg 10)
Hotspot Principal Components
hotspot.pcs = as.list(names(hotspot.genes))
names(hotspot.pcs) = names(hotspot.genes)
do.wave = pheno_clin[rownames(expr.mrna),"DOwave",drop=F]
Error in h(simpleError(msg, call)): error in evaluating the argument 'x' in selecting a method for function 'rownames': object 'expr.mrna' not found
wave.col = as.numeric(as.factor(do.wave[,1]))
Error in h(simpleError(msg, call)): error in evaluating the argument 'x' in selecting a method for function 'as.factor': object 'do.wave' not found
#hotspot.genes.rns <- list()
for(i in 1:length(hotspot.genes)) {
tmp = hotspot.genes[[i]] %>%
dplyr::select(starts_with("DO")) %>%
as.matrix() %>%
t() %>%
prcomp()
hotspot.pcs[[i]] = tmp$x
#rownames(hotspot.pcs[[i]]) <- gsub("\\.x","", rownames(hotspot.pcs[[i]]))
#rownames(hotspot.pcs[[i]]) <- gsub("\\.y","", rownames(hotspot.pcs[[i]]))
tmp = gather(data.frame(mouse = rownames(hotspot.pcs[[i]]), hotspot.pcs[[i]]), pc, value, -mouse)
tmp$mouse <- gsub("\\.x","", tmp$mouse)
tmp$mouse <- gsub("\\.y","", tmp$mouse)
tmp = left_join(tmp, pheno_clin %>% dplyr::select(mouse, sex, DOwave, diet_days), by = "mouse")
print(tmp %>%
filter(pc %in% paste0("PC", 1:4)) %>%
mutate(DOwave = factor(DOwave)) %>%
ggplot(aes(DOwave, value, fill = sex)) +
geom_boxplot() +
facet_grid(pc~.) +
labs(title = paste("Chr", names(hotspot.genes)[i], "Hotspot")))
}
Error in svd(x, nu = 0, nv = k): a dimension is zero
Key Points
.